
Sunday Science: A Pill To Treat Sickle Cell Disease? Compound That Activates Fetal Gene Raises New Hope
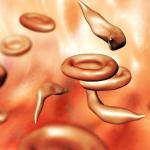
Last year, the U.S. Food and Drug Administration (FDA) approved two gene therapy procedures that can treat and, in some cases essentially cure sickle cell disease, a genetic blood disorder that causes pain and anemia in millions and still kills nearly 375,000 people worldwide every year. But the groundbreaking treatments require risky chemotherapy and cost some $2 million per person, putting them out of reach of the vast majority of sickle cell patients. Now, pharmaceutical researchers are reporting a potential oral drug that restores healthy blood cells in animal models of the disease.
Unlike a one-time gene therapy whose benefits could last decades, the new compound might have to be taken periodically for life and it hasn’t even begun safety testing in humans. But the experimental drug, described today in Science, offers hope that sickle cell disease could one day be widely and cheaply treatable with a simple pill.
“This seems promising,” says Lewis Hsu, a pediatric hematologist at the University of Illinois, Chicago, who wasn’t involved with the new research. However, he cautions, “there is a pipeline of sickle cell disease treatments that work in mice and cells that haven’t worked in people.”
Sickle cell disease is caused by a mutation in the gene responsible for making adult hemoglobin. The change causes normally disk-shaped red blood cells to adopt a characteristic crescent shape and to stick together, clogging capillaries, damaging tissues and triggering episodes of severe pain. One of the two newly approved therapies uses an engineered virus to insert copies of a modified version of the gene for adult hemoglobin into a patient’s own stem cells. Those programmed cells are then returned to the person after chemotherapy clears out existing blood stem cells.
The other approved therapy also modifies a patient’s blood stem cells but, uses the CRISPR gene-editing system to block a gene for a protein called BCL11A, which in adults represses the production of a form of hemoglobin used by fetuses. During infancy this fetal hemoglobin is normally dialed down as babies switch to making the adult version. So, when CRISPR turns off BCL11A’s gene, it restores production of fetal hemoglobin in the blood stem cells, helping sickle cell patients.
But the high cost and complexity of gene therapy has made it tough to see how either approach could work in lower income countries, especially those in Africa, where most people with sickle cell disease reside. “Regrettably, this therapy will not reach many patients,” says Jay Bradner, a sickle cell expert with the biopharmaceutical company Amgen.
Pharmaceutical companies have long been searching for drugs to switch on fetal hemoglobin in adults. In 1998, FDA approved a drug called hydroxyurea, which was found to modestly boost fetal hemoglobin production. But the drug can also suppress the proliferation of bone marrow stem cells, causing anemia and other side effects, a problem that has upended several other would-be sickle cell drugs as well. “The whole field has been struggling with this,” says Pamela Ting, a hematology researcher at Novartis.
A team led by Ting and Bradner has recently been searching for compounds that bind to a protein called cereblon, which is known to tag other proteins for destruction by cells’ natural protein recycling system. The original hope was they could find one that helped cereblon tag the BCL11A protein, thereby restoring fetal hemoglobin production. They screened a library of 2814 cereblon-binding molecules and fed those to immature red blood cells, unearthing one, called dWIZ-1, that increased fetal hemoglobin production.
But it unexpectedly didn’t target BCL11A, instead latching onto another gene regulatory protein called WIZ, which wasn’t known to be involved in controlling fetal hemoglobin levels. Further refinement of that compound produced dWIZ-2, which the team found raised fetal hemoglobin production in red blood cells from a baseline of 17% to 45%. The latter is at a level that would produce functional red blood cells if it translated into humans, Bradner says. The compound also worked when given orally to mice and in two out of three cynomolgus monkeys, all without apparent side effects, the team reports. “This is the first small molecule that induces fetal hemoglobin without damaging the stem cells,” Bradner says.
Despite this promise, the compound still faces challenges. For starters, WIZ is made in many cell types beyond red blood cells and appears to have a role in regulating the activity of numerous genes. “That could make it too broad an actor” to repress safely, says Stuart Orkin, a stem cell biologist at Harvard University’s Dana-Farber Cancer Institute and veteran sickle cell researcher not involved in the new work.
But Bradner notes that dWIZ-2 seems to have an outsized effect in boosting fetal hemoglobin. So, regardless of whether the Novartis experimental compound passes muster in clinical trials, it has already helped reveal a novel way to control which types of hemoglobin red blood cells churn out. And that, in and of itself, could set the stage for improved sickle cell drugs to come, Bradner suggests.
doi: 10.1126/science.z3iyub5
Bob Service is a news reporter for Science in Portland, Oregon, covering chemistry, materials science, and energy stories.
Science Magazine.
Support nonprofit science journalism
Help Science publish trustworthy, high-impact stories about research and the people who shape it. Subscribe to News from Science.

Nuclear Danger Is Growing. Physicists of the World, Unite!
Curtis T. Asplund, Zia Mian, Stewart Prager, Frank von Hippel
Bulletin of the Atomic Scientists
July 1, 2024